质谱法测定水中溶解氙的含量及其同位素组成
李军杰+刘汉彬 张佳+韩娟+金贵善+张建锋
摘要 利用设计的一套水样中提取并分离Xe的装置,与稀有气体质谱仪HelixSFT联用,建立了高精度水中溶解Xe的含量及其同位素组成的分析方法。对该装置的密封性及本底进行检测,证明此方法可靠;循环升温降温法分离Xe气中大量Ar的方法,可在短时间内除掉绝大部分Ar,减小其对Xe含量及同位素组成的测试影响。该方法的检出限可低至10
13~10
14L/L;对空气饱和水进行测试,Xe含量、129Xe/131Xe和132Xe/134Xe的测试结果的相对标准偏差分别为1.00%,0.10%和0.20%,误差分别为0.93%,0.16%和0.50%;分析了造成测试误差的因素,明确了误差的来源。
关键词水样;氙同位素;提取;质谱
1引言
自然界中Xe有9种稳定同位素:124Xe,126Xe,128Xe,129Xe,130Xe,131Xe,132Xe,134Xe和136Xe,核电站反应堆核裂变产生能量过程中,238U产生大量的132Xe和134Xe,235U产生大量的129Xe和131Xe[1\]。如果在初级循环水内这些同位素的含量明显升高,或者其同位素组成和大气Xe同位素组成有明显的差异,可能指示反应堆存在不同程度的核泄露[2~5\]。因此建立水中溶解Xe含量及同位素组成的测试方法有非常重要实际应用价值和意义。
水中溶解Xe含量及其同位素组成的测定主要包括水样采集、气体提取纯化以及气体分析等过程。目前国内样品采集方法主要是用两端带有玻璃阀门的玻璃管封装采集[6\],这种方法弊端在于阀门死角处极易形成气泡而干扰测定结果。国外采样方法主要用铜管采集法,使水样流经并充满铜管,最后用机械钳将铜管两端夹紧密封,其密封方式及铜管漏率极低,可以最大程度避免空气渗入而污染样品。但是需要谨慎操作,才能确保铜管两端被密封,另外,取样铜管连至系统后,在大气环境中打开铜管,操作不慎,会使铜管夹口壁薄处产生漏孔,导致大气渗入[7\]。气体提取纯化与分离是将水样引入真空系统,水中难溶气体析出,用吸气剂泵去除活性气体,在采用低温冷泵通过不断升温的方法在合适的温度下分离出Xe气进行以备测量[8~11\]。由于水中溶解Ar含量高,Ar在水中的溶解度比Kr和Xe高4个量级[9,12\],靠不断升温很难将其完全释放,会影响Xe的测定:其一,混合气体中的Ar显著稀释了待测Xe,使Xe的离子化效率降低,影响Xe测试的灵敏度,且导致Xe同位素测试的分馏\[9\];其二,Ar的压力相对较高,质谱仪灯丝寿命会缩短。Xe气的含量及同位素组成分析,主要使用四极杆质谱仪和稀有气体磁质谱仪两种仪器进行测量。四极杆质谱仪的优势在于高效、操作简单,磁质谱仪在灵敏度、分辨率等方面具有明显的优势,而大型磁质谱测量结果更加准确[13\]。
本研究建立了一种取样可靠、高效分离纯化样品中溶解Xe的方法。采用铜管取样,保证了气体的密封性;循环升温降温纯化分离Xe,实现了水中溶解Xe与其它气体的完全分离,利用稀有气体质谱仪HelixSFT对实验室内部标准水中溶解Xe含量和同位素组成进行了测试,获得了理想的测试结果。
2实验部分
2.1仪器与试剂
HelixSFT稀有气体质谱仪(美国ThermoFisher公司),配备Nier型离子源,飞行管道分叉设计,配备法拉第杯(Cup)及电子倍增器(SEM),其中电子倍增器配备偏转过滤电场,噪音值<10cpm,大大提高了测试结果的准确性,降低了仪器测试的检出限[14\];法拉第杯的分辨率达到400,电子倍增器的分辨率高达700,可以将不同Xe同位素完全分开。
试剂主要有干燥剂、吸附剂、无粉活性炭、ST101型吸气剂(ZrAl合金)、液氮、酒精干冰混合液,132Xe纯气、二次去离子水等。
2.2空气饱和水(ASW)的配制
去离子水在一定大气压力和温度下的溶解度是恒定的[7,13\],其同位素组成与大气中Xe同位素组成一致,将去离子水放置于大气压和温度恒定的空气中,使其充分溶解空气中的Xe,直至饱和,获得空气饱和水ASW(AirSaturatedWater),作为实验室内部的标准水。
本实验室将20L二次去离子水放置于室内环境温度为20℃、大气压力977Torr(130kPa)、通风良好的环境中,不断搅拌24h,使去离子水与空气充分交换平衡,将其封装待用。
2.3取样方法
采用外径为6.35mm,壁厚为0.9mm的无氧铜管取样,依次用酒精、去离子水、丙酮冲洗干净,60℃烘干待用。
取样时,将铜管竖直放置,水样通过微型水泵从铜管下端流经铜管并从上端排出,取样过程中连续敲击铜管赶走内部气泡,使得铜管内部全部充满水,用压力钳先把铜管上部夹断冷封,然后在距上部密封约30mm处,将底部夹断密封,放置待用。
2.4Xe氣提取分离
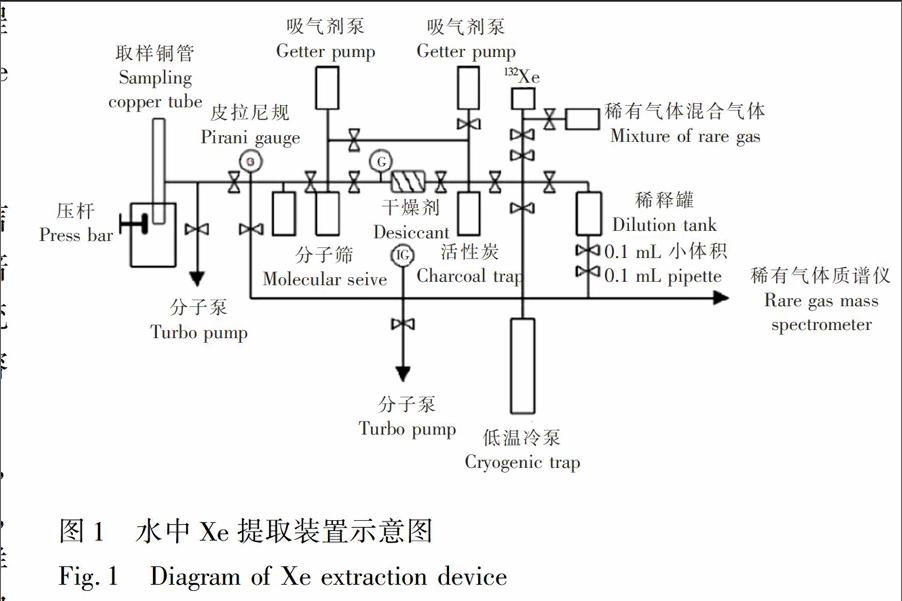
采集的水样中不仅溶解有He,Ne,Ar,Kr,Xe稀有气体,而且含有更多的N2,O2,CO2等活性气体,因此Xe的提取过程主要包括水中溶解气体的析出,析出气体中活性气体的吸附,稀有气体中Xe气的分离3个过程。本实验自主设计了一套JP水样Xe提取装置(图1),整套装置全部为金属连接,可在300℃高温条件下烘烤,真空泵组采用无油干泵,最大程度降低系统本底,整套系统真空度优于1.0×10
6Pa。
将装满空气饱和水样品的铜管用金属卡套密封连接至提取装置,将提取系统抽真空约1.0×10
6Pa,关闭真空泵阀门,高真空环境下压开铜管一端密封面,使水溶液扩散至一个约100mL的不锈钢罐内,用超声波振荡不锈钢罐底部,加速溶解气体析出,析出气体通过毛细管及冷阱,在冷阱和分子筛上套上液氮,将Ar,Kr和Xe以及少量水蒸汽冷凝,He和Ne则通过分子泵组抽走。将冷阱和分子筛加热至100℃,充分释放Ar,Kr,Xe以及少量的水蒸气,通过干燥剂阱充分除水,将其冷冻至干燥剂另一端的活性炭冷阱中,根据真空规检测除水程度,待干燥气体全部转移至活性炭冷阱后,加热活性炭冷阱至100℃,释放其中的气体,用锆铝泵充JP分吸附纯化其中的活性气体,低温冷泵温度调至60K,Ar、Kr和Xe冷冻至低温冷泵内,通过循环升温降温过程除去其中的Ar,将分离出的Kr和Xe引入质谱仪进行分析。
2.5质谱分析
优化Xe同位素测量参数,根据信号强度大小选用法拉第杯或者电子倍增器,用跳峰的方式依次测量整套系统本底值、大气Xe同位素组成和样品溶解Xe的同位素组成。
实验采用的标准气体为大气Xe,取0.1mL的空气样品引入纯化系统,采用与上述水样中Xe的纯化过程同样的步骤进行分离,用质谱进行测定。因大气Xe含量及同位素组成恒定,通过峰高比较法计算待测样品的Xe含量。测定空气中Xe同位素组成,并与实际值相比,用Linearlaw[15\]计算质量歧视因子,对水中测得溶解Xe同位素组成进行校正,得到水中溶解Xe同位素组成。
3结果与讨论
3.1取样铜管密封性
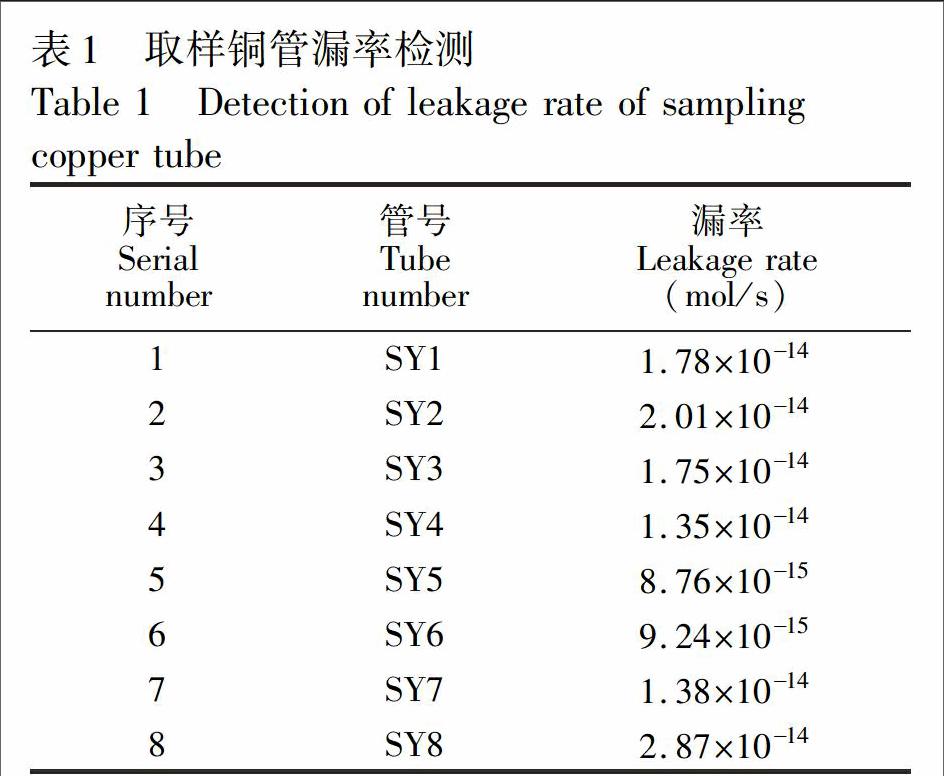
采用液压钳将已装满水样的铜管从两端夹断,利用铜管粘性进行冷封,将该铜管与氦质谱检漏仪连接,在其密封处利用氦气测试其漏率大小(表1),检测结果说明其密封性可靠。
Xe在20℃、1个标准大气压下的溶解度约为3.25×ZH(10
13mol/g,即10g水样中约有3.25×10
12mol的溶解Xe。空气中Xe的丰度约为8.7×10
8L/L[12\],按照铜管的最大漏率2.87×10
13mol/s计算,则空气中Xe在1个月内渗入到铜管量仅为6.47×10
15mol,造成的分析误差约0.2%,由于Xe的扩散能力比He更差,因此,Xe在铜管中由于漏率造成的误差小于0.2%。
3.2系统本底值
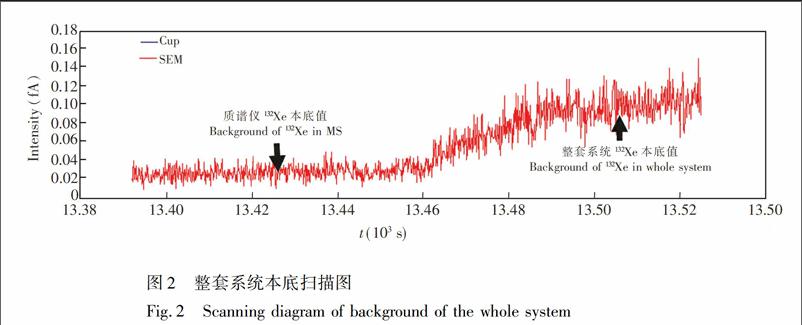
一个样品的提取、分离及测试过程需要1~2h,提取装置系统本身在这段时间内管壁或者密封焊接处自身会释放气体,因此,必须对整套系统的本底进行测试。选用大气Xe含量最高132Xe(26.9%)作为监测系统本ZH)底的指标。将取样铜管连入系统后,不打开铜管密封面,采用与样品提取及测试完全一致的步骤,对系统本底产生的132Xe进行扫描测定(图2)。
图2显示,质谱仪的本底约0.02fA,打开连接提取分离装置系统与质谱仪的进气阀门后,整套系统本底值约为0.10fA。对约10g空气饱和水中溶解Xe的测定,其132Xe的强度在3000fA左右,因此,本底值仅占实际信号强度0.0033%左右,整套装置系统的本底值对样品中Xe的测量可忽略不计。
3.3分离温度
稀有氣体He,Ne,Ar,Kr和Xe在空气饱和水中的含量大小为Ar>>Ne>Kr>He>Ne[16\],由于水中溶解的Ar的含量远高于Xe和其它稀有气体的含量,大量Ar不但会稀释Xe,导致Xe的测试重现性差,还会引起测试Xe过程中的同位素分馏效应。利用Ar与Xe在活性炭冷阱凝固点的差异,可对Ar进行分离移除。引入0.1mL纯Ar气,调节装有活性炭冷阱的低温冷泵至60K,将其完全冻入冷阱内,逐渐升温,通过质谱仪信号大小确定了其释放温度曲线。引入0.1mL纯132Xe,采用和纯Ar同样的步骤,确定Xe的释放温度曲线(图3)。
由图3可知,在150K温度下,可以将Ar充分释放,而Xe几乎依然保留在活性炭冷阱内,实验过程中分别引入Ar和132Xe纯气,且两者信号强度几乎一致的情况下而获得的释放曲线。但在实际样品测量过程中,Ar和Xe是以混合气体的形式存在的,即使在60K时将Ar,Kr和Xe全部转移至低温冷阱内,再升至150K,释放并用分子泵抽走Ar,发现Ar信号强度依然很高。Stanley[17\]指出,这可能是因为Xe的原子半径比较大,虽然含量较低,在低温条件下,“包裹”住大量Ar原子而使其难以释放,实验采用了不断升温降温的循环释放气体的办法,即先关闭真空泵组阀门,将低温冷阱升至325K,充分释放Ar,Kr和Xe,再降温至150K,冷冻住Xe,打开泵组阀门,抽走在冷阱上部的Ar,然后循环此过程4~5次,相比于单一的150K冷冻Xe而抽走Ar的方式,可以更高效分离掉其中大量的Ar气(图4)。
DZ(K1
本实验每个样品循环测定20次,由于稀有气体质谱仪采用静态模式下测量,因此每次循环测定的结果均按回归曲线计算零点回归值,然后计算每个样品在90%置信水平下的置信区间。
由表2可知,Xe含量、129Xe/131Xe和132Xe/134Xe的测定值的相对标准偏差分别为1.00%,0.10%和0.20%,而其相对误差分别为0.93%,0.16%和0.50%,说明水中溶解Xe的含量及其同位素组成的测量,结果重现性好,准确度高。
3.5方法的检出限
方法的检出限与仪器的灵敏度、噪音值以及方法的本底值稳定性有关。仪器的灵敏度越高,噪音值越低,整套系统本底值越低且稳定,则该方法的检出限越低[14\]。灵敏度和噪音值是由质谱仪的性能来决定的,与其离子源、聚焦透镜及检测器等有关。本实验所采用的HelixSFT稀有气体质谱仪灵敏度极高,对于Xe,可以达到1.0×10
5A/Pa,而接收器采用1012Ω高阻法拉第杯及电子倍增器,电子倍增器的噪音值<10cpm,对于极低的信号,可以实现准确测量,由图5和图6可知,对于较低丰度的124Xe和126Xe同位素,法拉第杯受限于噪音值而不能够检测到其信号,但电子倍增器可以很好地检测到其信号。DZ(K1
如前所述,整套系统的132Xe本底值非常低。根据质谱仪对Xe的测试灵敏度计算,取约10g空气饱和水样品,132Xe可以产生3000fA左右的信号。分析未知Xe含量的样品的测试误差小于1%范围内,根据系统本底的不确定度,如果水样中132Xe产生大于5fA的信号强度,则样品的本底对测试结果造成的误差小于1%,通过与空气饱和水中132Xe产生的强度比较,可知产生5fA的信号强度水中Xe的含量可以低至7.3×10
14L/L。因此,本方法对水中溶解Xe浓度低至10
13~10
14L/L的水样,在一定误差范围内可以准确测定。
3.6误差分析
为了对水中溶解Xe含量和同位素组成测定的准确度进一步评估,分析了误差的主要来源(表3)。
6[18\],存在1.10%的不确定度。取空气的体积也存在0.40%的误差,而水中溶解Xe的含量是由一定量空气中Xe含量校正得到,因此会导致结果有1.30%的测试误差。但由于一定温度和压力下空气饱和水中Xe含量的理论计算值也是由空气中Xe的含量计算而得,因此,由于空气中Xe含量的不确定性引起的误差可能会减小甚至消除[8,11,19\]。JP
其次,稀有气体在水中的溶解度受温度、压力的影响,尤其对于Xe,其溶解度对温度的变化最为敏感,温度每升高1℃,其溶解度变化可以达到4.2%[20\],因此在空气饱和水制备的过程中,压力计和温度计对于周围环境大气压力和温度的测量会带来0.12%的误差。
Xe在从水中提取分离的过程中,难以将其中的溶解Xe全部提取完成[8\],在Xe与其它气体分离的过程中,由于不断升温降温的反复循环过程,即使在Xe完全冷凝的温度下,依然可以在电子倍增器上检测到略高于本底水平且稳定的Xe计数,图7为132Xe的时间扫描图,活性炭冷阱全部释放Xe后,132Xe的强度约为3000fA,在150K时,依然有约3fA的132Xe气体,可能是冷阱存在一定的温阶,极少量的Xe吸附在冷阱上端管壁不断释放引起的,该过程造成0.10%的误差。
最后,部分误差可能源自取样铜管本身的漏率及铜管本身的释气过程,该误差随着取样铜管放置时间的延长而变大。在保证取样铜管密封良好的情况下,应尽早分析样品,减小误差。
水中溶解Xe同位素组成测试过程中的误差主要源于空气中Xe同位素组成的不确定性,即使仪器存在质量歧视效应,经过大气Xe同位素组成的测试,可获得相应的校正因子,对水样中溶解Xe同位素组成测测试结果进行校正,可减小由此带来的测试误差[21~23\]。有研究者称不同的气体含量会对本身的同位素组成造成一定的影响,即压力歧视效应[24\],目前尚没有这方面具体的研究报道。与水中溶解Xe含量的测定相比,水中Xe同位素组成的测定的影响因素明显减少,其误差值要远低于水中Xe含量测定的误差值。
4结论
建立了一种水中溶解Xe的提取分离、含量與同位素组成测试的方法,整套系统具有低漏率、低本底、低检出限等优点。循环升温降温分离Xe中Ar的方法,可高效分离其中的Ar。对空气饱和水中溶解Xe的含量和同位素组成测定,结果重现性好、准确度高。分析了测试过程中的误差来源,提出了准确分析水样中溶解Xe的含量及同位素组成过程中值得注意的影响因素。
References
1(#PalcsuL,SzantoZS,SvingorE,MolnarM,FutoI,MajorZ.InternationalConference,NuclearEnergyforNewEurope,2002,0702.10702.7
2ZHANGXiaoZhi,LIUDaMing,LIJinYing,LIAnLi,JINXiaoHai.JournalofNuclearandRadiochemistry.2004,26(4),doi:02539950(2004)04019806
张小枝,刘大鸣,李金英,李安利,金小海.HTK核化学与放射化学,2004,26(4),doi:02539950(2004)04019806
3AregbeY,ValkiersS,MayerK,VarlamM,WellumR.EsardaBulletin.,2007,37:30-34
4AregbeY,MayerK,ValkiersS,BievreD.J.Anal.Chem.,1997,358:533-535
5MolnarM,PalcsuL,MajorZ.J.RadioanalNucl.Chem.,2010,doi:10.1007/s1096701007921
6ZHANGXiaoBao,XUYongChang,CHENJianPing,SUNMingLiang,TUJianQi,LIXiuFen.ActaPetroleiSinica,2004,25(2):41-46
张晓宝,徐永昌,陈建平,孙明良,涂建淇,李秀芬.HTK石油学报,2004,25(2):41-46
7JasonCP,McNeilG,LangmanSR,DennisF.Appl.Geochem.,1997,12:707-714
8HammeRC,EmersonSR.Mar.Chem.,2004,91:53-64LM
9BeyerleU,AeschbachW,ImbodenDM,BaurH,GrafT,KipferR.Environ.Sci.Technol.,2000,34:2042-2054
10AeschbachHertigW,PeetersF,BeyerleU,KipferR.WaterResour.Res.,1999,35(9):2779-2792
11WeissRF.J.Chem.Engineer.Data,1971,16:235-241
12WANGXianBin.GeochemistryandCosmochemistryofNobleGasIsotope.,Beijing:SciencePress,1989:66-68
王先彬.HTK稀有气体同位素地球化学和宇宙化学.北京:科学出版社1989:66-68
13SanoY,TakahataN.J.Oceanogr.,2005,61(3):465-473
14BurnardP,ZimmermannL,SanoY.TheNobleGasesasGeochemicalTracers,2013:1-15
15RennePR,SharpWD,DienoAL.QuarternGeochronol.,2009,4(4):288-298
16HuntAG.U.S.GeologicalSurveyTechniquesandMethods,2015,book5,chap.A11,p22
17StanleyRHR,BaschekB,LottDE,JenkinsWJ.Geochem.Geophys.Geosystems,2009,10(5),Q05008
18OzimaM,PodpsekFA.NobleGasGeochemistry,2nd.2002
19HammeRC,EmersonSR.Mar.Chem.,2004,91(14):53-64
20KesterDR.ChemicalOceanography,AcademicPress,NewYork,1975:498-556
21ValkiersS,SchaeferF,DeBievre.SeparationTechnology,ElsevierScienceB.V.,1994:965-968
22OzimaM,PodpsekFA.NobleGasGeochem.,2nd.2002
23BeyerleU,AeschbachW,ImbodenDM,BaurH,GrafT,KipferR.Environ.Sci.Technol,2000,34:2042-2054
24BurnardPG,FarleyKA.Geochem.Geophys.Geosystems.,2000,1:1022)
AbstractAmethodwasestablishedtoanalyzethedissolvedXeconcentrationandXeisotopesinwaterpreciselywiththeselfdesigndeviceofXeextractionandseparationlineincombiningwiththeHelixSFTnoblegasmassspectrometer.Theleaktestandthebackgrounddeterminationofthewholesystemprovedthesystemwasreliable.ThelargeamountofArgonfromXecouldbeeffectivelyremovedbycyclingtemperaturerisingandfallingmethod,whichresultedintheeliminationoftheinterferencetotheanalysisofXeconcentrationandisotopes.ThelimitofdetectionofthemethodforXeconcentrationcouldreachaslowas10
13-10
14L/L.ThemethodwasusedforthemeasurementofthedissolvedXeinairsaturatedwater(ASW),andtheRSDsofXeconcentration,129Xe/131Xeand132Xe/134Xewere1.0%,0.10%and0.20%respectively,whiletheerrorswere0.93%,0.16%and0.50%respectively.
KeywordsWatersample;Xenonisotopes;Extraction;Massspectrometry
HQWT6JY(Received4April2016;accepted9August2016)
摘要 利用设计的一套水样中提取并分离Xe的装置,与稀有气体质谱仪HelixSFT联用,建立了高精度水中溶解Xe的含量及其同位素组成的分析方法。对该装置的密封性及本底进行检测,证明此方法可靠;循环升温降温法分离Xe气中大量Ar的方法,可在短时间内除掉绝大部分Ar,减小其对Xe含量及同位素组成的测试影响。该方法的检出限可低至10
13~10
14L/L;对空气饱和水进行测试,Xe含量、129Xe/131Xe和132Xe/134Xe的测试结果的相对标准偏差分别为1.00%,0.10%和0.20%,误差分别为0.93%,0.16%和0.50%;分析了造成测试误差的因素,明确了误差的来源。
关键词水样;氙同位素;提取;质谱
1引言
自然界中Xe有9种稳定同位素:124Xe,126Xe,128Xe,129Xe,130Xe,131Xe,132Xe,134Xe和136Xe,核电站反应堆核裂变产生能量过程中,238U产生大量的132Xe和134Xe,235U产生大量的129Xe和131Xe[1\]。如果在初级循环水内这些同位素的含量明显升高,或者其同位素组成和大气Xe同位素组成有明显的差异,可能指示反应堆存在不同程度的核泄露[2~5\]。因此建立水中溶解Xe含量及同位素组成的测试方法有非常重要实际应用价值和意义。
水中溶解Xe含量及其同位素组成的测定主要包括水样采集、气体提取纯化以及气体分析等过程。目前国内样品采集方法主要是用两端带有玻璃阀门的玻璃管封装采集[6\],这种方法弊端在于阀门死角处极易形成气泡而干扰测定结果。国外采样方法主要用铜管采集法,使水样流经并充满铜管,最后用机械钳将铜管两端夹紧密封,其密封方式及铜管漏率极低,可以最大程度避免空气渗入而污染样品。但是需要谨慎操作,才能确保铜管两端被密封,另外,取样铜管连至系统后,在大气环境中打开铜管,操作不慎,会使铜管夹口壁薄处产生漏孔,导致大气渗入[7\]。气体提取纯化与分离是将水样引入真空系统,水中难溶气体析出,用吸气剂泵去除活性气体,在采用低温冷泵通过不断升温的方法在合适的温度下分离出Xe气进行以备测量[8~11\]。由于水中溶解Ar含量高,Ar在水中的溶解度比Kr和Xe高4个量级[9,12\],靠不断升温很难将其完全释放,会影响Xe的测定:其一,混合气体中的Ar显著稀释了待测Xe,使Xe的离子化效率降低,影响Xe测试的灵敏度,且导致Xe同位素测试的分馏\[9\];其二,Ar的压力相对较高,质谱仪灯丝寿命会缩短。Xe气的含量及同位素组成分析,主要使用四极杆质谱仪和稀有气体磁质谱仪两种仪器进行测量。四极杆质谱仪的优势在于高效、操作简单,磁质谱仪在灵敏度、分辨率等方面具有明显的优势,而大型磁质谱测量结果更加准确[13\]。
本研究建立了一种取样可靠、高效分离纯化样品中溶解Xe的方法。采用铜管取样,保证了气体的密封性;循环升温降温纯化分离Xe,实现了水中溶解Xe与其它气体的完全分离,利用稀有气体质谱仪HelixSFT对实验室内部标准水中溶解Xe含量和同位素组成进行了测试,获得了理想的测试结果。
2实验部分
2.1仪器与试剂
HelixSFT稀有气体质谱仪(美国ThermoFisher公司),配备Nier型离子源,飞行管道分叉设计,配备法拉第杯(Cup)及电子倍增器(SEM),其中电子倍增器配备偏转过滤电场,噪音值<10cpm,大大提高了测试结果的准确性,降低了仪器测试的检出限[14\];法拉第杯的分辨率达到400,电子倍增器的分辨率高达700,可以将不同Xe同位素完全分开。
试剂主要有干燥剂、吸附剂、无粉活性炭、ST101型吸气剂(ZrAl合金)、液氮、酒精干冰混合液,132Xe纯气、二次去离子水等。
2.2空气饱和水(ASW)的配制
去离子水在一定大气压力和温度下的溶解度是恒定的[7,13\],其同位素组成与大气中Xe同位素组成一致,将去离子水放置于大气压和温度恒定的空气中,使其充分溶解空气中的Xe,直至饱和,获得空气饱和水ASW(AirSaturatedWater),作为实验室内部的标准水。
本实验室将20L二次去离子水放置于室内环境温度为20℃、大气压力977Torr(130kPa)、通风良好的环境中,不断搅拌24h,使去离子水与空气充分交换平衡,将其封装待用。
2.3取样方法
采用外径为6.35mm,壁厚为0.9mm的无氧铜管取样,依次用酒精、去离子水、丙酮冲洗干净,60℃烘干待用。
取样时,将铜管竖直放置,水样通过微型水泵从铜管下端流经铜管并从上端排出,取样过程中连续敲击铜管赶走内部气泡,使得铜管内部全部充满水,用压力钳先把铜管上部夹断冷封,然后在距上部密封约30mm处,将底部夹断密封,放置待用。
2.4Xe氣提取分离
采集的水样中不仅溶解有He,Ne,Ar,Kr,Xe稀有气体,而且含有更多的N2,O2,CO2等活性气体,因此Xe的提取过程主要包括水中溶解气体的析出,析出气体中活性气体的吸附,稀有气体中Xe气的分离3个过程。本实验自主设计了一套JP水样Xe提取装置(图1),整套装置全部为金属连接,可在300℃高温条件下烘烤,真空泵组采用无油干泵,最大程度降低系统本底,整套系统真空度优于1.0×10
6Pa。
将装满空气饱和水样品的铜管用金属卡套密封连接至提取装置,将提取系统抽真空约1.0×10
6Pa,关闭真空泵阀门,高真空环境下压开铜管一端密封面,使水溶液扩散至一个约100mL的不锈钢罐内,用超声波振荡不锈钢罐底部,加速溶解气体析出,析出气体通过毛细管及冷阱,在冷阱和分子筛上套上液氮,将Ar,Kr和Xe以及少量水蒸汽冷凝,He和Ne则通过分子泵组抽走。将冷阱和分子筛加热至100℃,充分释放Ar,Kr,Xe以及少量的水蒸气,通过干燥剂阱充分除水,将其冷冻至干燥剂另一端的活性炭冷阱中,根据真空规检测除水程度,待干燥气体全部转移至活性炭冷阱后,加热活性炭冷阱至100℃,释放其中的气体,用锆铝泵充JP分吸附纯化其中的活性气体,低温冷泵温度调至60K,Ar、Kr和Xe冷冻至低温冷泵内,通过循环升温降温过程除去其中的Ar,将分离出的Kr和Xe引入质谱仪进行分析。
2.5质谱分析
优化Xe同位素测量参数,根据信号强度大小选用法拉第杯或者电子倍增器,用跳峰的方式依次测量整套系统本底值、大气Xe同位素组成和样品溶解Xe的同位素组成。
实验采用的标准气体为大气Xe,取0.1mL的空气样品引入纯化系统,采用与上述水样中Xe的纯化过程同样的步骤进行分离,用质谱进行测定。因大气Xe含量及同位素组成恒定,通过峰高比较法计算待测样品的Xe含量。测定空气中Xe同位素组成,并与实际值相比,用Linearlaw[15\]计算质量歧视因子,对水中测得溶解Xe同位素组成进行校正,得到水中溶解Xe同位素组成。
3结果与讨论
3.1取样铜管密封性
采用液压钳将已装满水样的铜管从两端夹断,利用铜管粘性进行冷封,将该铜管与氦质谱检漏仪连接,在其密封处利用氦气测试其漏率大小(表1),检测结果说明其密封性可靠。
Xe在20℃、1个标准大气压下的溶解度约为3.25×ZH(10
13mol/g,即10g水样中约有3.25×10
12mol的溶解Xe。空气中Xe的丰度约为8.7×10
8L/L[12\],按照铜管的最大漏率2.87×10
13mol/s计算,则空气中Xe在1个月内渗入到铜管量仅为6.47×10
15mol,造成的分析误差约0.2%,由于Xe的扩散能力比He更差,因此,Xe在铜管中由于漏率造成的误差小于0.2%。
3.2系统本底值
一个样品的提取、分离及测试过程需要1~2h,提取装置系统本身在这段时间内管壁或者密封焊接处自身会释放气体,因此,必须对整套系统的本底进行测试。选用大气Xe含量最高132Xe(26.9%)作为监测系统本ZH)底的指标。将取样铜管连入系统后,不打开铜管密封面,采用与样品提取及测试完全一致的步骤,对系统本底产生的132Xe进行扫描测定(图2)。
图2显示,质谱仪的本底约0.02fA,打开连接提取分离装置系统与质谱仪的进气阀门后,整套系统本底值约为0.10fA。对约10g空气饱和水中溶解Xe的测定,其132Xe的强度在3000fA左右,因此,本底值仅占实际信号强度0.0033%左右,整套装置系统的本底值对样品中Xe的测量可忽略不计。
3.3分离温度
稀有氣体He,Ne,Ar,Kr和Xe在空气饱和水中的含量大小为Ar>>Ne>Kr>He>Ne[16\],由于水中溶解的Ar的含量远高于Xe和其它稀有气体的含量,大量Ar不但会稀释Xe,导致Xe的测试重现性差,还会引起测试Xe过程中的同位素分馏效应。利用Ar与Xe在活性炭冷阱凝固点的差异,可对Ar进行分离移除。引入0.1mL纯Ar气,调节装有活性炭冷阱的低温冷泵至60K,将其完全冻入冷阱内,逐渐升温,通过质谱仪信号大小确定了其释放温度曲线。引入0.1mL纯132Xe,采用和纯Ar同样的步骤,确定Xe的释放温度曲线(图3)。
由图3可知,在150K温度下,可以将Ar充分释放,而Xe几乎依然保留在活性炭冷阱内,实验过程中分别引入Ar和132Xe纯气,且两者信号强度几乎一致的情况下而获得的释放曲线。但在实际样品测量过程中,Ar和Xe是以混合气体的形式存在的,即使在60K时将Ar,Kr和Xe全部转移至低温冷阱内,再升至150K,释放并用分子泵抽走Ar,发现Ar信号强度依然很高。Stanley[17\]指出,这可能是因为Xe的原子半径比较大,虽然含量较低,在低温条件下,“包裹”住大量Ar原子而使其难以释放,实验采用了不断升温降温的循环释放气体的办法,即先关闭真空泵组阀门,将低温冷阱升至325K,充分释放Ar,Kr和Xe,再降温至150K,冷冻住Xe,打开泵组阀门,抽走在冷阱上部的Ar,然后循环此过程4~5次,相比于单一的150K冷冻Xe而抽走Ar的方式,可以更高效分离掉其中大量的Ar气(图4)。
DZ(K1
本实验每个样品循环测定20次,由于稀有气体质谱仪采用静态模式下测量,因此每次循环测定的结果均按回归曲线计算零点回归值,然后计算每个样品在90%置信水平下的置信区间。
由表2可知,Xe含量、129Xe/131Xe和132Xe/134Xe的测定值的相对标准偏差分别为1.00%,0.10%和0.20%,而其相对误差分别为0.93%,0.16%和0.50%,说明水中溶解Xe的含量及其同位素组成的测量,结果重现性好,准确度高。
3.5方法的检出限
方法的检出限与仪器的灵敏度、噪音值以及方法的本底值稳定性有关。仪器的灵敏度越高,噪音值越低,整套系统本底值越低且稳定,则该方法的检出限越低[14\]。灵敏度和噪音值是由质谱仪的性能来决定的,与其离子源、聚焦透镜及检测器等有关。本实验所采用的HelixSFT稀有气体质谱仪灵敏度极高,对于Xe,可以达到1.0×10
5A/Pa,而接收器采用1012Ω高阻法拉第杯及电子倍增器,电子倍增器的噪音值<10cpm,对于极低的信号,可以实现准确测量,由图5和图6可知,对于较低丰度的124Xe和126Xe同位素,法拉第杯受限于噪音值而不能够检测到其信号,但电子倍增器可以很好地检测到其信号。DZ(K1
如前所述,整套系统的132Xe本底值非常低。根据质谱仪对Xe的测试灵敏度计算,取约10g空气饱和水样品,132Xe可以产生3000fA左右的信号。分析未知Xe含量的样品的测试误差小于1%范围内,根据系统本底的不确定度,如果水样中132Xe产生大于5fA的信号强度,则样品的本底对测试结果造成的误差小于1%,通过与空气饱和水中132Xe产生的强度比较,可知产生5fA的信号强度水中Xe的含量可以低至7.3×10
14L/L。因此,本方法对水中溶解Xe浓度低至10
13~10
14L/L的水样,在一定误差范围内可以准确测定。
3.6误差分析
为了对水中溶解Xe含量和同位素组成测定的准确度进一步评估,分析了误差的主要来源(表3)。
6[18\],存在1.10%的不确定度。取空气的体积也存在0.40%的误差,而水中溶解Xe的含量是由一定量空气中Xe含量校正得到,因此会导致结果有1.30%的测试误差。但由于一定温度和压力下空气饱和水中Xe含量的理论计算值也是由空气中Xe的含量计算而得,因此,由于空气中Xe含量的不确定性引起的误差可能会减小甚至消除[8,11,19\]。JP
其次,稀有气体在水中的溶解度受温度、压力的影响,尤其对于Xe,其溶解度对温度的变化最为敏感,温度每升高1℃,其溶解度变化可以达到4.2%[20\],因此在空气饱和水制备的过程中,压力计和温度计对于周围环境大气压力和温度的测量会带来0.12%的误差。
Xe在从水中提取分离的过程中,难以将其中的溶解Xe全部提取完成[8\],在Xe与其它气体分离的过程中,由于不断升温降温的反复循环过程,即使在Xe完全冷凝的温度下,依然可以在电子倍增器上检测到略高于本底水平且稳定的Xe计数,图7为132Xe的时间扫描图,活性炭冷阱全部释放Xe后,132Xe的强度约为3000fA,在150K时,依然有约3fA的132Xe气体,可能是冷阱存在一定的温阶,极少量的Xe吸附在冷阱上端管壁不断释放引起的,该过程造成0.10%的误差。
最后,部分误差可能源自取样铜管本身的漏率及铜管本身的释气过程,该误差随着取样铜管放置时间的延长而变大。在保证取样铜管密封良好的情况下,应尽早分析样品,减小误差。
水中溶解Xe同位素组成测试过程中的误差主要源于空气中Xe同位素组成的不确定性,即使仪器存在质量歧视效应,经过大气Xe同位素组成的测试,可获得相应的校正因子,对水样中溶解Xe同位素组成测测试结果进行校正,可减小由此带来的测试误差[21~23\]。有研究者称不同的气体含量会对本身的同位素组成造成一定的影响,即压力歧视效应[24\],目前尚没有这方面具体的研究报道。与水中溶解Xe含量的测定相比,水中Xe同位素组成的测定的影响因素明显减少,其误差值要远低于水中Xe含量测定的误差值。
4结论
建立了一种水中溶解Xe的提取分离、含量與同位素组成测试的方法,整套系统具有低漏率、低本底、低检出限等优点。循环升温降温分离Xe中Ar的方法,可高效分离其中的Ar。对空气饱和水中溶解Xe的含量和同位素组成测定,结果重现性好、准确度高。分析了测试过程中的误差来源,提出了准确分析水样中溶解Xe的含量及同位素组成过程中值得注意的影响因素。
References
1(#PalcsuL,SzantoZS,SvingorE,MolnarM,FutoI,MajorZ.InternationalConference,NuclearEnergyforNewEurope,2002,0702.10702.7
2ZHANGXiaoZhi,LIUDaMing,LIJinYing,LIAnLi,JINXiaoHai.JournalofNuclearandRadiochemistry.2004,26(4),doi:02539950(2004)04019806
张小枝,刘大鸣,李金英,李安利,金小海.HTK核化学与放射化学,2004,26(4),doi:02539950(2004)04019806
3AregbeY,ValkiersS,MayerK,VarlamM,WellumR.EsardaBulletin.,2007,37:30-34
4AregbeY,MayerK,ValkiersS,BievreD.J.Anal.Chem.,1997,358:533-535
5MolnarM,PalcsuL,MajorZ.J.RadioanalNucl.Chem.,2010,doi:10.1007/s1096701007921
6ZHANGXiaoBao,XUYongChang,CHENJianPing,SUNMingLiang,TUJianQi,LIXiuFen.ActaPetroleiSinica,2004,25(2):41-46
张晓宝,徐永昌,陈建平,孙明良,涂建淇,李秀芬.HTK石油学报,2004,25(2):41-46
7JasonCP,McNeilG,LangmanSR,DennisF.Appl.Geochem.,1997,12:707-714
8HammeRC,EmersonSR.Mar.Chem.,2004,91:53-64LM
9BeyerleU,AeschbachW,ImbodenDM,BaurH,GrafT,KipferR.Environ.Sci.Technol.,2000,34:2042-2054
10AeschbachHertigW,PeetersF,BeyerleU,KipferR.WaterResour.Res.,1999,35(9):2779-2792
11WeissRF.J.Chem.Engineer.Data,1971,16:235-241
12WANGXianBin.GeochemistryandCosmochemistryofNobleGasIsotope.,Beijing:SciencePress,1989:66-68
王先彬.HTK稀有气体同位素地球化学和宇宙化学.北京:科学出版社1989:66-68
13SanoY,TakahataN.J.Oceanogr.,2005,61(3):465-473
14BurnardP,ZimmermannL,SanoY.TheNobleGasesasGeochemicalTracers,2013:1-15
15RennePR,SharpWD,DienoAL.QuarternGeochronol.,2009,4(4):288-298
16HuntAG.U.S.GeologicalSurveyTechniquesandMethods,2015,book5,chap.A11,p22
17StanleyRHR,BaschekB,LottDE,JenkinsWJ.Geochem.Geophys.Geosystems,2009,10(5),Q05008
18OzimaM,PodpsekFA.NobleGasGeochemistry,2nd.2002
19HammeRC,EmersonSR.Mar.Chem.,2004,91(14):53-64
20KesterDR.ChemicalOceanography,AcademicPress,NewYork,1975:498-556
21ValkiersS,SchaeferF,DeBievre.SeparationTechnology,ElsevierScienceB.V.,1994:965-968
22OzimaM,PodpsekFA.NobleGasGeochem.,2nd.2002
23BeyerleU,AeschbachW,ImbodenDM,BaurH,GrafT,KipferR.Environ.Sci.Technol,2000,34:2042-2054
24BurnardPG,FarleyKA.Geochem.Geophys.Geosystems.,2000,1:1022)
AbstractAmethodwasestablishedtoanalyzethedissolvedXeconcentrationandXeisotopesinwaterpreciselywiththeselfdesigndeviceofXeextractionandseparationlineincombiningwiththeHelixSFTnoblegasmassspectrometer.Theleaktestandthebackgrounddeterminationofthewholesystemprovedthesystemwasreliable.ThelargeamountofArgonfromXecouldbeeffectivelyremovedbycyclingtemperaturerisingandfallingmethod,whichresultedintheeliminationoftheinterferencetotheanalysisofXeconcentrationandisotopes.ThelimitofdetectionofthemethodforXeconcentrationcouldreachaslowas10
13-10
14L/L.ThemethodwasusedforthemeasurementofthedissolvedXeinairsaturatedwater(ASW),andtheRSDsofXeconcentration,129Xe/131Xeand132Xe/134Xewere1.0%,0.10%and0.20%respectively,whiletheerrorswere0.93%,0.16%and0.50%respectively.
KeywordsWatersample;Xenonisotopes;Extraction;Massspectrometry
HQWT6JY(Received4April2016;accepted9August2016)
